Biopython
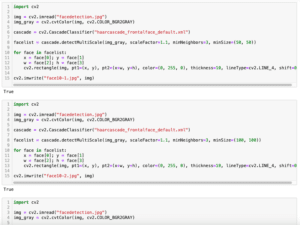
前回、openCVで顔、笑顔、目を検出する方法を紹介しました。
今回からは一応大きな括りでは専門分野であるバイオ系のライブラリ「Biopython」を紹介していきます。
ただバイオ系の研究者ではあるものの、バイオインフォマティクスの研究者ではないのでそこのところ注意してください。
バイオ系の研究者が、独学でPythonを学んで、それでもってBiopythonをちょっといじってみようというレベルのお話です。
それでも一人でも多くの人のためになればと思います。
それでは始めていきましょう。
Biopythonとは?
BiopythonとはPythonで書かれた無料で使用できるバイオ系のライブラリのことです。
バイオでは遺伝子配列やタンパク質配列など、文字列としての情報を多く扱いますし、タンパク質構造なんかは文字列だけではなく、3次元座標情報などを扱います。
遺伝子組み換え実験では、得られた情報をさらに加工して、新しい配列情報にしたものを設計図として、実際の実験に役立てたりします。
そのような遺伝子情報やタンパク質情報を扱うバイオ系のデータベースはたくさんあり、それぞれによって記述方法が違ったりします。
Biopythonではそのようにそれぞれ違う記述方式を読み込み、同じコマンドで呼び出せたり、またその後の情報の加工ができたりとバイオ系の研究に大きく貢献してくれるライブラリです。
ということでバイオ系の研究をしていて、Pythonを勉強しているのなら、触っておきたいライブラリかなと思います。
インストールは他のライブラリと同じくpipを使ってインストールできます。
pip install biopython配列の格納
まずは手打ちで配列を準備し、Biopythonに認識させてみましょう。
Biopythonに配列を認識させるためには「Bio.Seq内のSeqモジュール」を使います。
from Bio.seq import Seqそして配列を認識させ、変数に格納するには「Seq(“配列”)」を用います。
from Bio.Seq import Seq
DNA_seq = Seq("GGATCCGAATTC")
print(DNA_seq)
実行結果
GGATCCGAATTCちなみにこのように変数に格納した配列の型は「Bio.SeqのSeq型」となります。
from Bio.Seq import Seq
DNA_seq = Seq("GGATCCGAATTC")
print(type(DNA_seq))
実行結果
<class 'Bio.Seq.Seq'>ちなみにDNA配列だけでなく、RNAでもタンパク質でも格納できます。
from Bio.Seq import Seq
RNA_seq = Seq("GGAUCCGAAUUC")
print(RNA_seq)
実行結果
GGAUCCGAAUUCfrom Bio.Seq import Seq
Protein_seq = Seq("MATSELYKCPQDVN")
print(Protein_seq)
実行結果
MATSELYKCPQDVNさらにDNA、RNA、タンパク質で使われない文字でも特にエラーになることはありません。
from Bio.Seq import Seq
unknown_seq = Seq("OJZXB")
print(unknown_seq)
実行結果
OJZXB配列の種類に関して
上述のようにSeq型はDNA、RNA、タンパク質のどれであるかは認識していないようです。
逆に認識させることができるのかを調べてみました。
そこで見つけたのがこちらのページ。
「Bio.Alphabet」の「IUPACモジュール」を使うと配列がDNAなのか、RNAなのか、タンパク質なのかの情報を組み込めるようです。
ということで試してみましょう。
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
DNA_seq = Seq("GGATCCGAATTC", IUPAC.ambiguous_dna)
print(DNA_seq)
print(DNA_seq.alphabet)
実行結果
---------------------------------------------------------------------------
ImportError Traceback (most recent call last)
Cell In[24], line 2
1 from Bio.Seq import Seq
----> 2 from Bio.Alphabet import IUPAC
4 DNA_seq = Seq("GGATCCGAATTC", IUPAC.ambiguous_dna)
6 print(DNA_seq)
File /Library/Frameworks/Python.framework/Versions/3.10/lib/python3.10/site-packages/Bio/Alphabet/__init__.py:20
1 # Copyright 2000-2002 by Andrew Dalke.
2 # Revisions copyright 2007-2010 by Peter Cock.
3 # All rights reserved.
(...)
7 # Please see the LICENSE file that should have been included as part of this
8 # package.
9 """Alphabets were previously used to declare sequence type and letters (OBSOLETE).
10
11 The design of Bio.Aphabet included a number of historic design choices
(...)
17 transition from Bio.Alphabet to molecule type annotations.
18 """
---> 20 raise ImportError(
21 "Bio.Alphabet has been removed from Biopython. In many cases, the alphabet can simply be ignored and removed from scripts. In a few cases, you may need to specify the ``molecule_type`` as an annotation on a SeqRecord for your script to work correctly. Please see https://biopython.org/wiki/Alphabet for more information."
22 )
ImportError: Bio.Alphabet has been removed from Biopython. In many
cases, the alphabet can simply be ignored and removed from scripts.
In a few cases, you may need to specify the ``molecule_type`` as an
annotation on a SeqRecord for your script to work correctly. Please
see https://biopython.org/wiki/Alphabet for more information.「Bio.Alphabet has been removed from Biopython.」ということで、どうやらBio.Alphabetはもう存在しないようです(2023年7月時点 Biopython ver. 1.81)。
この「Bio.Alphabet」を使った情報は結構出てきますので、ご注意ください。
ただ単純に無くなっただけで、配列の種類を組み込めないわけではありません。
こちらのページにBio.Alphabetが亡くなったことへの対処をどうしたら良いか書かれています。
このページによると「SeqRecord」という関数を使うと配列にその配列の種類の情報を組み込めるようです。
その場合、まずは「from Bio.SeqRecord import SeqRecord」でSeqRecordモジュールをインポートします。
そして「SeqRecord(配列, id=”配列のID”, name=”配列の名前”, annotations={“molecule_type”: “種類”}, description=”概要”)」でその配列の情報をまとめることができます。
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
DNA_seq = Seq("GGATCCGAATTC")
DNA_record = SeqRecord(DNA_seq, id="DNA_test", name="DNA1", annotations={"molecule_type": "DNA"}, description="New DNA")
RNA_seq = Seq("GGAUCCGAAUUC")
RNA_record = SeqRecord(DNA_seq, id="RNA_test", name="RNA1", annotations={"molecule_type": "RNA"}, description="New RNA")
Protein_seq = Seq("MATSELYKCPQDVN")
Protein_record = SeqRecord(Protein_seq, id="Protein_test", name="Protein1", annotations={"molecule_type": "Protein"}, description="New Protein")
print(DNA_record)
print(RNA_record)
print(Protein_record)
実行結果
ID: DNA_test
Name: DNA1
Description: New DNA
Number of features: 0
/molecule_type=DNA
Seq('GGATCCGAATTC')
ID: RNA_test
Name: RNA1
Description: New RNA
Number of features: 0
/molecule_type=RNA
Seq('GGATCCGAATTC')
ID: Protein_test
Name: Protein1
Description: New Protein
Number of features: 0
/molecule_type=Protein
Seq('MATSELYKCPQDVN')ただし配列の種類(DNAか、RNAか、タンパク質か)を指定したとしても、特に配列がチェックされて特定の文字しか使えなくなるというわけではないようです。
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
DNA_seq = Seq("MATSELYKCPQDVN")
DNA_record = SeqRecord(DNA_seq, id="DNA_test", name="DNA1", annotations={"molecule_type": "DNA"}, description="New DNA")
print(DNA_record)
実行結果
ID: DNA_test
Name: DNA1
Description: New DNA
Number of features: 0
/molecule_type=DNA
Seq('MATSELYKCPQDVN')ということでどの変数にどんな配列を格納したかはしっかり把握しておく必要がありそうです。
次回はデータベースから配列をダウンロードしてBiopythonで読み込んでみます。
ではでは今回はこんな感じで。
コメント