Biopython
前回、バイオインフォマティクス用ライブラリBiopythonのインストールと配列の取り扱い方法を紹介しました。
今回はfasta形式とembl形式のファイルの読み込みとデータ取得の方法を紹介します。
それでは始めていきましょう。
Uniprotでの配列データのダウンロード
まずは読み込むための配列データをダウンロードしましょう。
配列データの取得にはNCBIを使われる方が多いと思いますが、個人的にはUniprotが好きなので、今回はUniprotでデータをダウンロードします。
Uniprotにアクセスしたら、中央の検索窓に検索したい遺伝子やタンパク質名を入れて「Search」をクリックします。
今回はとりあえずバイオ系なら誰もが知っているくらい有名なタンパク質である緑色蛍光タンパク質(Green Fluorescent Protein:GFP)としました。
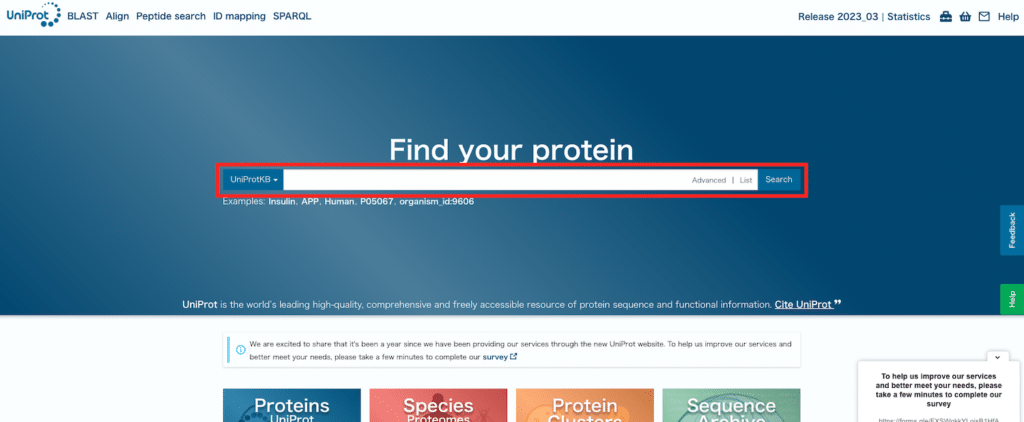
表示形式を「Cards」か「Table」かを選択するウインドウが出ます。
どうやら最近(2023年7月くらい?)変わったようで、私はこれまで慣れ親しんできた「Table」形式を選択しています。
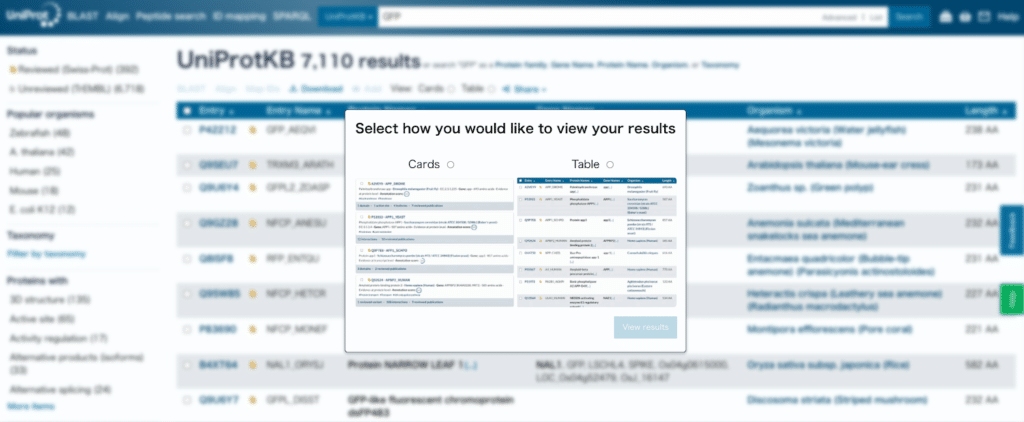
検索結果が表示されますので、目的の遺伝子、タンパク質の一番左の列にあるEntryのIDをクリックします。
GFPの場合はP42212です。
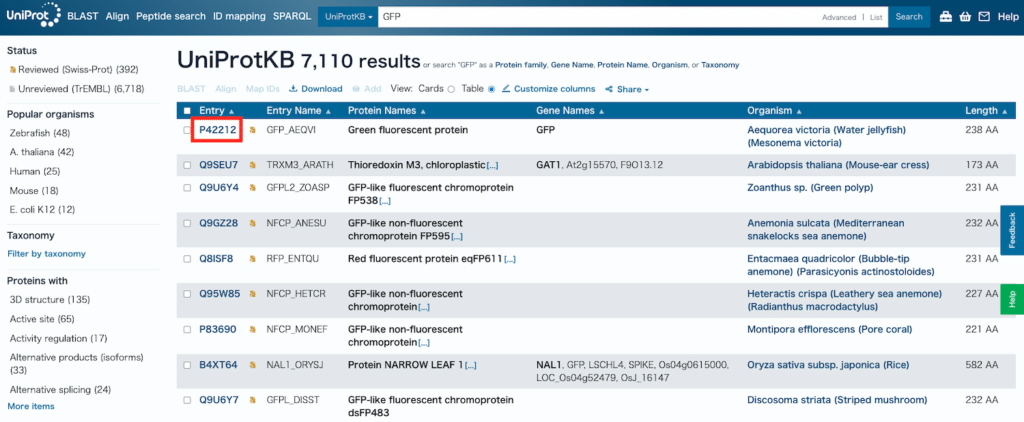
選択した遺伝子、タンパク質の情報がずらっと出てきますので、左の目次から「Sequence」をクリックします。
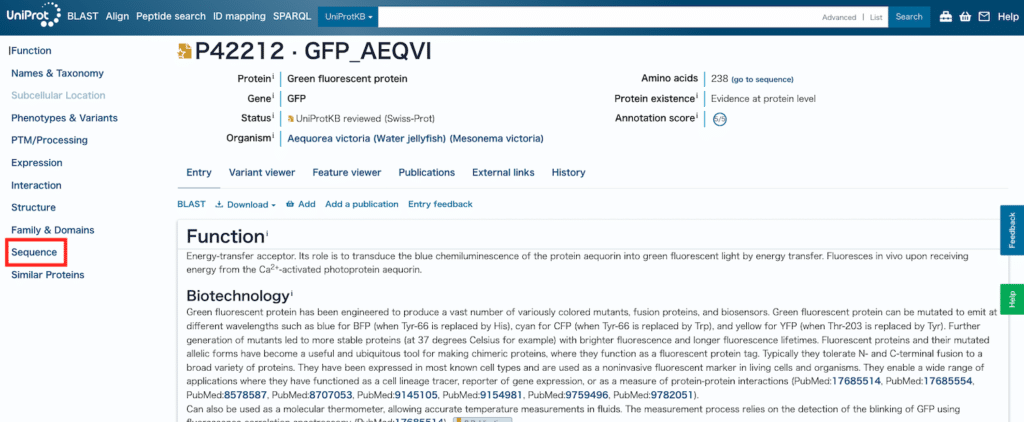
少し下にスクロールします。
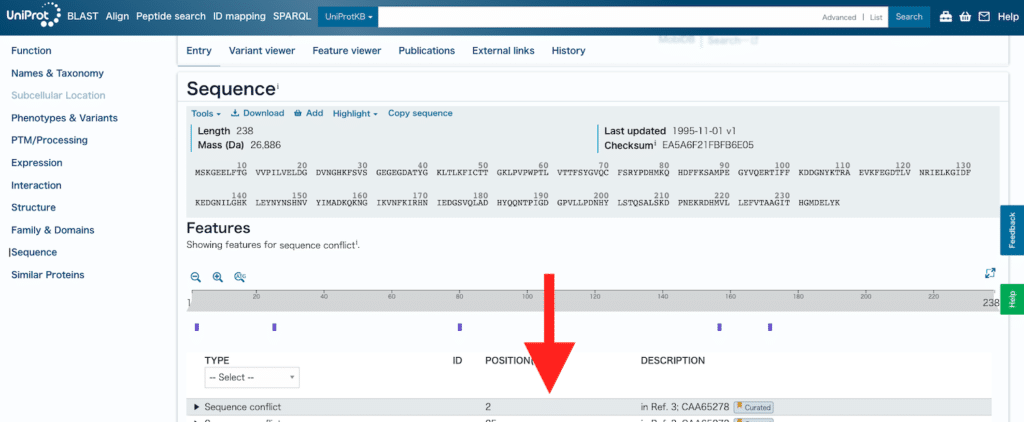
「Sequence database」で目的の遺伝子、タンパク質のリンクをクリックします。
今回は「AAA27722.1」のEMBLをクリックしました。
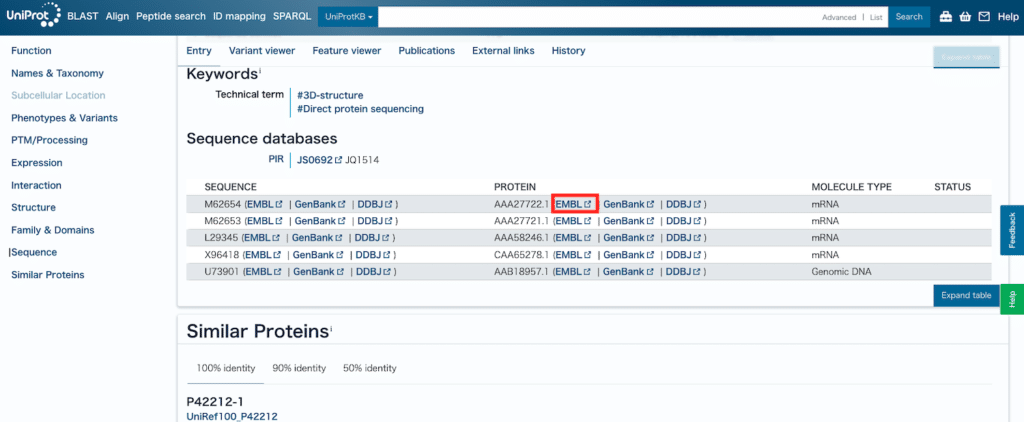
EMBLのデータベースに飛びますので、右の「Download」から「EMBL」と「FASTA」の両方をダウンロードします、
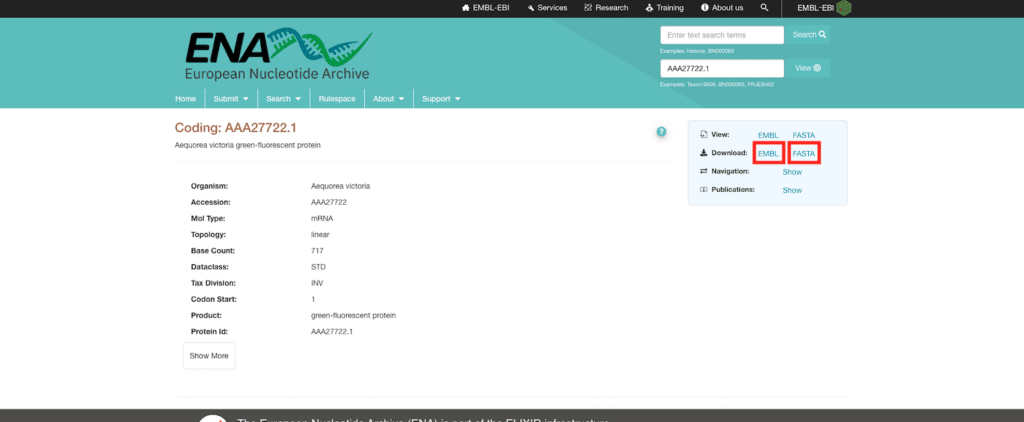
これで目的のファイルのダウンロード完了です。
Biopythonでのファイルの読み込みとデータの取得
それではBiopythonでファイルを読み込み、その中のデータを取得してみましょう。
まずはfastaファイルの読み込みを試していきます。
ファイルの読み込みには「SeqIO」モジュールを用い、「SeqIO.parse(”fastaファイルのパス”, “fasta”)」とします。
そして取り出す際の注意ですが、SeqIOはFASTA形式としてはmulti-FASTA形式で扱っているようで、複数のfastaデータが含まれていると認識しています。
また格納されている形式が特殊なようで、それぞれのデータの取り出しにはfor文を使って一つずつデータを取り出す必要があります。
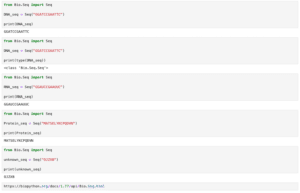
from Bio import SeqIO
record = SeqIO.parse("AAA27722.1.fasta", "fasta")
for content in record:
print(content)
実行結果
ID: ENA|AAA27722|AAA27722.1
Name: ENA|AAA27722|AAA27722.1
Description: ENA|AAA27722|AAA27722.1 Aequorea victoria green-fluorescent protein
Number of features: 0
Seq('ATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTA...TAA')一つのデータに含まれるそれぞれの情報を取得するには、上の例だとこのようになります。
- ID:content.id
- 配列名:content.name
- 概要:content.description
- 特徴:content.geatures
- 配列:content.seq
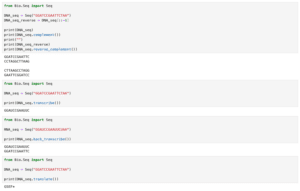
from Bio import SeqIO
record = SeqIO.parse("AAA27722.1.fasta", "fasta")
for content in record:
print(content.id)
print(content.name)
print(content.description)
print(content.features)
print(content.seq)
実行結果
ENA|AAA27722|AAA27722.1
ENA|AAA27722|AAA27722.1
ENA|AAA27722|AAA27722.1 Aequorea victoria green-fluorescent protein
[]
ATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAA
TGGGCACAAATTCTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACATACGGAAAACTTACCCTTAAAT
TTATTTGCACTACTGGAAAGCTACCTGTTCCATGGCCAACACTTGTCACTACTTTCTCTTATGGTGTT
CAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAGAGTGCCATGCCCGAAGG
TTATGTACAGGAAAGAACTATATTTTACAAAGATGACGGGAACTACAAATCACGTGCTGAAGTCAAGT
TTGAAGGTGATACCCTCGTTAATAGAATTGAGTTAAAAGGTATTGATTTTAAAGAAGATGGAAACATT
CTTGGACACAAAATGGAATACAACTATAACTCACACAATGTATACATCATGGCAGACAAACAAAAGAA
TGGAATCAAAGTTAACTTCAAAATTAGACACAACATTGAAGATGGAAGCGTTCAACTAGCAGACCATT
ATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCCACACAA
TCTGCCCTTTCCAAAGATCCCAACGAAAAGAGAGATCACATGATCCTTCTTGAGTTTGTAACAGCTGC
TGGGATTACACATGGCATGGATGAACTATACAAATAAちなみにファイル読み込みとfor文でのデータ取得を合わせてしまうことも可能です。
from Bio import SeqIO
for content in SeqIO.parse("AAA27722.1.fasta", "fasta"):
print(content.id)
print(content.name)
print(content.description)
print(content.features)
print(content.seq)
実行結果
ENA|AAA27722|AAA27722.1
ENA|AAA27722|AAA27722.1
ENA|AAA27722|AAA27722.1 Aequorea victoria green-fluorescent protein
[]
ATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAA
TGGGCACAAATTCTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACATACGGAAAACTTACCCTTAAAT
TTATTTGCACTACTGGAAAGCTACCTGTTCCATGGCCAACACTTGTCACTACTTTCTCTTATGGTGTT
CAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAGAGTGCCATGCCCGAAGG
TTATGTACAGGAAAGAACTATATTTTACAAAGATGACGGGAACTACAAATCACGTGCTGAAGTCAAGT
TTGAAGGTGATACCCTCGTTAATAGAATTGAGTTAAAAGGTATTGATTTTAAAGAAGATGGAAACATT
CTTGGACACAAAATGGAATACAACTATAACTCACACAATGTATACATCATGGCAGACAAACAAAAGAA
TGGAATCAAAGTTAACTTCAAAATTAGACACAACATTGAAGATGGAAGCGTTCAACTAGCAGACCATT
ATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCCACACAA
TCTGCCCTTTCCAAAGATCCCAACGAAAAGAGAGATCACATGATCCTTCTTGAGTTTGTAACAGCTGC
TGGGATTACACATGGCATGGATGAACTATACAAATAAEMBL形式のファイルを読み込むには「SeqIO.parse(“EMBL形式のファイルパス”, “embl”)」とします。
from Bio import SeqIO
for content in SeqIO.parse("AAA27722.1.txt", "embl"):
print(content.id)
print(content.name)
print(content.description)
print(content.features)
print(content.seq)
実行結果
AAA27722.1
AAA27722
Aequorea victoria green-fluorescent protein
[SeqFeature(SimpleLocation(ExactPosition(0), ExactPosition(717),
strand=1), type='source', qualifiers=...), SeqFeature(CompoundLocation
([SimpleLocation(ExactPosition(207), ExactPosition(413), strand=1,
ref='M62654.1'), SimpleLocation(ExactPosition(945), ExactPosition(1240),
strand=1, ref='M62654.1'), SimpleLocation(ExactPosition(2307),
ExactPosition(2523), strand=1, ref='M62654.1')], 'join'), type='CDS',
location_operator='join', qualifiers=...)]
ATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAA
TGGGCACAAATTCTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACATACGGAAAACTTACCCTTAAAT
TTATTTGCACTACTGGAAAGCTACCTGTTCCATGGCCAACACTTGTCACTACTTTCTCTTATGGTGTT
CAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAGAGTGCCATGCCCGAAGG
TTATGTACAGGAAAGAACTATATTTTACAAAGATGACGGGAACTACAAATCACGTGCTGAAGTCAAGT
TTGAAGGTGATACCCTCGTTAATAGAATTGAGTTAAAAGGTATTGATTTTAAAGAAGATGGAAACATT
CTTGGACACAAAATGGAATACAACTATAACTCACACAATGTATACATCATGGCAGACAAACAAAAGAA
TGGAATCAAAGTTAACTTCAAAATTAGACACAACATTGAAGATGGAAGCGTTCAACTAGCAGACCATT
ATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCCACACAA
TCTGCCCTTTCCAAAGATCCCAACGAAAAGAGAGATCACATGATCCTTCTTGAGTTTGTAACAGCTGC
TGGGATTACACATGGCATGGATGAACTATACAAATAAちなみにSeqIOで読み込める形式はかなり多く、シークエンスのファイル形式である「abi」やアライメントで有名な「clustal」、Swiss-Prot形式である「swiss」なども読み込めるようです。
こちらに一覧があるのでよかったらどうぞ。
次回は相補鎖、反転配列、逆相補鎖、転写前後の配列、翻訳後の配列の作成方法を紹介します。
ではでは今回はこんな感じで。
コメント