Biopython
前回、Biopythonでコドン表の表示と各生物のコドン表を使った翻訳方法を紹介しました。
今回は2本の遺伝子やタンパク質の配列を比較するペアワイズアライメントの方法を紹介します。
色々みていくうちに「Alignモジュール」を使う方法と「pairwais2モジュール」を使う方法を見つけたのでそれぞれ見てみましょう。
それでは始めていきましょう。
Alignモジュールを使う方法
まずはAlignモジュールを使う方法です。
「from Bio import Align」でモジュールをインポートします。
そして「aligner = Align.PairwiseAligner(match_score=スコア)」でインスタンスを作成し、「aligner.align(配列1, 配列2)」でアライメントを行います。
結果は複数得られるので、for文を使って一つずつ取り出します。
from Bio import Align
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
aligner = Align.PairwiseAligner(match_score=1.0)
alignments = aligner.align(DNA_seq1, DNA_seq2)
for alignment in alignments:
print(alignment)
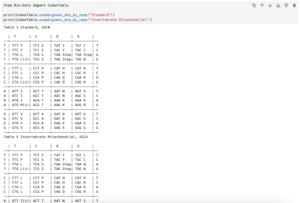
実行結果
target 0 GAATTCGAATTC 12
0 -|-|-||----- 12
query 0 -A-T-CG----- 4
target 0 GAATTCGAATTC 12
0 --||-||----- 12
query 0 --AT-CG----- 4
target 0 GAATTCGAATTC 12
0 -|--|||----- 12
query 0 -A--TCG----- 4
target 0 GAATTCGAATTC 12
0 --|-|||----- 12
query 0 --A-TCG----- 4「Align.PairwiseAligner」の場合、基本的には「グローバルアライメント」となり、2つの配列を全体で合わせようとします。
「ローカルアライメント」、つまり部分的にマッチする部分を探す場合は「aligner.mode = “local”」を追加します。
from Bio import Align
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
aligner = Align.PairwiseAligner(match_score=1.0)
aligner.mode = "local"
alignments = aligner.align(DNA_seq1, DNA_seq2)
for alignment in alignments:
print(alignment)
実行結果
target 1 AATTCG 7
0 |-|-|| 6
query 0 A-T-CG 4
target 2 ATTCG 7
0 ||-|| 5
query 0 AT-CG 4
target 1 AATTCG 7
0 |--||| 6
query 0 A--TCG 4
target 2 ATTCG 7
0 |-||| 5
query 0 A-TCG 4アライメントのスコアを表示する場合は「aligner.score(配列1, 配列2)」を用います。
from Bio import Align
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
aligner = Align.PairwiseAligner(match_score=1.0)
score = aligner.score(DNA_seq1, DNA_seq2)
print(score)
実行結果
4.0ちなみに前回のコドン表を変えて翻訳した配列比較の場合はこうなります。
from Bio import SeqIO, Align
for record in SeqIO.parse("AAA27722.1.fasta", "fasta"):
DNA_seq =record.seq
default_seq = DNA_seq.translate()
im_seq = DNA_seq.translate(table="Invertebrate Mitochondrial")
aligner = Align.PairwiseAligner(match_score=1.0)
alignments = aligner.align(default_seq, im_seq)
for alignment in alignments:
print(alignment)
実行結果
target 0 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
query 0 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
target 60 VTTFSYGVQCFSR-YPDHMKQHDFFKSAMPEGYVQER-TI-FYKDDGNYKSRAEVKFEGD
60 ||||||||||||--||||||||||||||||||||||--|--|||||||||||||||||||
query 60 VTTFSYGVQCFS-SYPDHMKQHDFFKSAMPEGYVQE-ST-MFYKDDGNYKSRAEVKFEGD
target 117 TLVNR-IELKGIDFKEDGNILGHKMEYNYNSHNVYIMADKQKNGIKVNFKIR-HNIEDGS
120 ||||--|||||||||||||||||||||||||||||||||||||||||||||--|||||||
query 117 TLVN-SIELKGIDFKEDGNILGHKMEYNYNSHNVYIMADKQKNGIKVNFKI-SHNIEDGS
target 175 VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKR-DHMILLEFVTAAGITHGMD
180 |||||||||||||||||||||||||||||||||||||||--|||||||||||||||||||
query 175 VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEK-SDHMILLEFVTAAGITHGMD
target 234 ELYK* 239
240 ||||| 245
query 234 ELYK* 239
target 0 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
query 0 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
target 60 VTTFSYGVQCFS-RYPDHMKQHDFFKSAMPEGYVQER-TI-FYKDDGNYKSRAEVKFEGD
60 ||||||||||||--||||||||||||||||||||||--|--|||||||||||||||||||
query 60 VTTFSYGVQCFSS-YPDHMKQHDFFKSAMPEGYVQE-ST-MFYKDDGNYKSRAEVKFEGD
target 117 TLVNR-IELKGIDFKEDGNILGHKMEYNYNSHNVYIMADKQKNGIKVNFKIR-HNIEDGS
120 ||||--|||||||||||||||||||||||||||||||||||||||||||||--|||||||
query 117 TLVN-SIELKGIDFKEDGNILGHKMEYNYNSHNVYIMADKQKNGIKVNFKI-SHNIEDGS
target 175 VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKR-DHMILLEFVTAAGITHGMD
180 |||||||||||||||||||||||||||||||||||||||--|||||||||||||||||||
query 175 VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEK-SDHMILLEFVTAAGITHGMD
target 234 ELYK* 239
240 ||||| 245
query 234 ELYK* 239
(以下略)pairwise2モジュールを使う方法
ペアワイズアライメントをするにはもう一つ「pairwise2モジュール」を使う方法があります。
この場合、「from Bio import pairwise2」で「pairwise2モジュール」をインポートし、「pairwise2.align.globalxx(配列1, 配列2)」でグローバルアライメントを行います。
from Bio import pairwise2
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
results = pairwise2.align.globalxx(DNA_seq1, DNA_seq2)
for result in results:
print(result)
実行結果
Alignment(seqA='GAATTCGAATTC', seqB='--A-TCG-----', score=4.0, start=0, end=12)
Alignment(seqA='GAATTCGAATTC', seqB='-A--TCG-----', score=4.0, start=0, end=12)
Alignment(seqA='GAATTCGAATTC', seqB='--AT-CG-----', score=4.0, start=0, end=12)
Alignment(seqA='GAATTCGAATTC', seqB='-A-T-CG-----', score=4.0, start=0, end=12)ローカルアライメントの場合は「pairwise2.align.localxx(配列1, 配列2)」となります。
from Bio import pairwise2
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
results = pairwise2.align.localxx(DNA_seq1, DNA_seq2)
for result in results:
print(result)
実行結果
Alignment(seqA='GAATTCGAATTC', seqB='--A-TCG-----', score=4.0, start=2, end=7)
Alignment(seqA='GAATTCGAATTC', seqB='-A--TCG-----', score=4.0, start=1, end=7)
Alignment(seqA='GAATTCGAATTC', seqB='--AT-CG-----', score=4.0, start=2, end=7)
Alignment(seqA='GAATTCGAATTC', seqB='-A-T-CG-----', score=4.0, start=1, end=7)このままでは見づらいので表示のフォーマットを変更します。
「from Bio.pairwise2 import format_alignment」で「format_alignmentモジュール」をインポートします。
そして結果を表示する際に「format_alignment(*結果)」とします。
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
results = pairwise2.align.globalxx(DNA_seq1, DNA_seq2)
for result in results:
print(format_alignment(*result))
実行結果
GAATTCGAATTC
| |||
--A-TCG-----
Score=4
GAATTCGAATTC
| |||
-A--TCG-----
Score=4
GAATTCGAATTC
|| ||
--AT-CG-----
Score=4
GAATTCGAATTC
| | ||
-A-T-CG-----
Score=4ちなみに変数の前に「*」がついています(*result)が、これは変数がリストであり、アンパックをしています。
また「gloablxx」、「localxx」と最後に「xx」がついていますが、これらを変えることでマッチのパラメータやギャップのパラメータを変更することができます。
詳しくはこちらのページをどうぞ(ただしBiopythonのバージョンが古いので注意)。
例えば「pairwise2.align.globalms(配列1, 配列2, マッチ, ミスマッチ, ギャップ開始, ギャップ伸長)」という形で用いる。
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
DNA_seq1 = "GAATTCGAATTC"
DNA_seq2 = "ATCG"
results = pairwise2.align.globalms(DNA_seq1, DNA_seq2, 2, -1, -0.5, -0.1)
for result in results:
print(format_alignment(*result))
実行結果
GAATTCGAATTC
| |||
--A-TCG-----
Score=6
GAATTCGAATTC
| |||
-A--TCG-----
Score=6
GAATTCGAATTC
|| ||
--AT-CG-----
Score=6これでとりあえず配列比較まではできるようになったので、一旦Biopythonはここまでにします。
また何かやりたいことが出てきたら、色々試して紹介します。
次回はPandasで外れ値を除外したり、置換したりする方法を紹介します。
ではでは今回はこんな感じで。
コメント